
 Wunde, Wundheilung
Wunde, Wundheilung Infektion
Infektion Akutes Abdomen
Akutes Abdomen Abdominaltrauma
Abdominaltrauma Ileus
Ileus Hernien
Hernien- Allgemeine Chirurgie
 Struma benigna
Struma benigna Schilddrüsen-CA
Schilddrüsen-CA Nebenschilddrüsen
Nebenschilddrüsen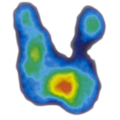 Hypderthyreose
Hypderthyreose Nebenniere
Nebenniere- Endokrine Chirurgie
 Achalasie
Achalasie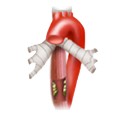 Ösophagus-CA
Ösophagus-CA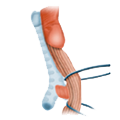 Ösophagusdivertikel
Ösophagusdivertikel Ösophagusperforation
Ösophagusperforation Verätzung
Verätzung Magen-CA
Magen-CA Ulkuskrankheit
Ulkuskrankheit GERD
GERD Adipositas
Adipositas- Oberer GI-Trakt
 CED
CED Divertikulitis
Divertikulitis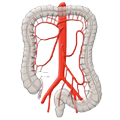 Kolon-CA
Kolon-CA Proktologie
Proktologie Rektum-CA
Rektum-CA- Unterer GI-Trakt
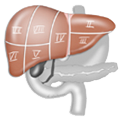 Aantomie
Aantomie Ikterus
Ikterus Cholezystolithiais
Cholezystolithiais Benigne Leberläsionen
Benigne Leberläsionen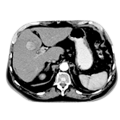 Maligne Leberläsionen
Maligne Leberläsionen Pankreatitis
Pankreatitis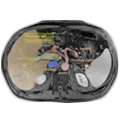 Pankreaskarzinom
Pankreaskarzinom- Hepatobiliäre Chirurgie
Anatomie
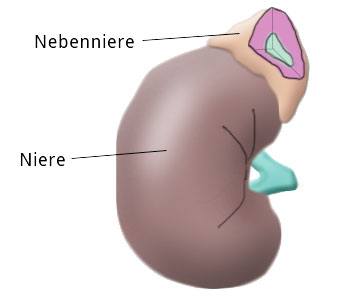
Die Nebennieren liegen den Nieren an ihrem kranialen Pol an und haben ansonsten mit diesem Organ nicht viel gemeinsam.
Funktionell-anatomisch gliedert sich die Nebenniere in das Nebennierenmark und die Nebennierenrinde, die histologisch weiter in die Zona glomerulosa, Zona fasciculata und Zona reticularis unterteilt werden kann.
In der Zona glomerulosa werden Mineralokortikoide wie das Aldosteron produziert, in der Zona fasciculata Glukokortikoide wie Cortison und Cortisol und in der Zona reticularis die Androgene.
Das Nebennierenmark ist die Produktionsstätte von Adrenalin, Noradrenalin und Dopamin.
Phäochromozytom
Symptomatik
Eine Nebennierenmarküberfuntion geht mit einer übersteigerten Produktion und Ausschüttung von Adrenalin und Noradrenalin einher. Verbunden ist dies sehr oft mit einer arteriellen Hypertonie, die sich medikamentös nur schlecht einstellen lässt. Unruhe, gesteigerte Erregbarkeit und generelle Stoffwechselerhöhung sind weitere Symptome.
Ein Phäochromozytom kann familiär gehäuft auftreten im Zusammenhang mit sog. multiplen endokrinen Neoplasien, vor allem mit der MEN II in Kombination mit einem medulären Schilddrüsenkarzinom und einem Hyperparathyreoidismus.
Lokalisation
In der Mehrzahl der Fälle ist ein Phäochromozytom in den Nebennieren lokalisiert, es kann aber in 10-15% der Fälle auch eine extraadrenale Lokalisation vorkommen. Eine adrenale Manifestation bedeutet aber nicht automatisch, dass der Befund nur einseitig vorliegen muss. Auch bilaterale Befunde sind in ca. 5% möglich, sollten dann aber an das Vorliegen eines MEN II denken lassen.
Diagnostik
Neben der Anamnese, die wichtige Hinweise auf das Krankheitsbild liefern kann, spielt die Labordiagnostik eine entscheidende Rolle beim Erkennen eines Phäochromozytoms. Im Blut werden die Werte für Adrenalin und Noradrenalin bestimmt. Im 24-h Sammelurin kann man deren Abbaustoffe, das Metanephrin und die Vanilin-Mandelsäure bestimmen. Als Tumormarker dienen Chromogranin A und Dopamin, ein erhöhtes Calcitonin kann auf ein MEN hinweisen.
Zur Lokalisation eines Phäochromozytoms kann eine Computertomografie oder MRT des Adbomens eingesetzt werden. Eventuell kann eine selektive Blutentnahme helfen, eine fragliche Seitenlokalisation zu klären.
Auch eine Szintigrafie mit Methyl-123-Jod-Benzy-Guanidin (MIBG) ist möglich und weist eine hohe Spezifität für das Vorliegen eines Phäochromozytoms auf.
Therapie
Mit der Diagnose steht auch die Operationsindikation. Bei einem Phäochromozytom besteht die Gefahr, bei intraoperativer Manipulation durch Freisetzung großer Mengen von Katecholaminen eine hypertensive Krise auszulösen. Dies kann zum Einen durch sorgfältiges Präparieren verhindert werden, essentiell wichtig ist bei einem Phäochromozytom allerdings eine Vorbehandlung mit einem Alpha-Blocker und Betablocker. Diese Vorbehandlung wird über einen Zeitraum von 10-14 Tagen durchgeführt und verhindert ein übermäßiges Ausschütten von Adrenalin und Noradrenalin während der Operation.
Prinzipiell steh ein transabdomineller oder ein retroperitonealer Zugang über einen Flankenschnitt zur Verfügung. Auch laparoskopisch lassen sich die Nebennieren gut erreichen. Es ist somit eine Operation mit minimalem Operationstrauma möglich.
Cushing-Syndrom
Eine Überproduktion von Glukokortikoiden lässt auf ein Cushing-Syndrom oder Morbus Cushing schließen. Die Ursache kann dabei in der Nebennierenrinde liegen (adrenaler Cushing) oder in der Hypophyse (zentraler Cushing).
Besser unterscheiden kann man diese Formen durch den Einfluss des ACTH. Wird dieses vermehrt in der Hypophyse gebildet, z.B. durch ein Hypophysenadenom, wird dadurch die Nebenniere vermehrt stimuliert. Man spricht von einer ACTH-bedingten oder sekundären Form des Cushings. Hat das ACTH keinen Einfluss, liegt die Störung in der Nebennierenrinde. Dies kann ebenfalls ein Adenom oder ein Karzinom sein, man bezeichnet diese Form als ACTH-unabhängig oder als primäre Form.
Differentialdiagnostisch kommt noch ein Cushinoid infrage, eine durch exogene Zufuhr verursachte Überschreitung der Cushing-Schwelle.
Adrenaler Morbus Cushing
präoperativ



Anamnese & Diagnostik
Patienten mit einem Morbus Cuhsing berichten über eine Gewichtszunahme und weisen typische morphologische Veränderungen auf. Dies können z.B. ein sog. Vollmondgesicht sein, Stammfettsucht, ein Stiernacken und in typischer Weise rot gefärbte Striae distensae rubrae. Auch eine arterielle Hypertonie kommt häufig vor. In der Labordiagnostik fällt ein erhöhter Plasmaspiegel für Kortison und ggf. ACTH auf.
Mit dem sog. Dexamethason-Hemmtest wird versucht, zwischen der primären und sekundären Form des M. Cushing zu unterscheiden. Zur Lokalisation dienen wiederum CT und MRT, ggf. seitengetrennte Blutentnahme und Hormonbestimmung. Auch Osteoporose und Störung der Libido oder Pontenz sind möglich. Bei Frauen stellen sich in einigen Fällen Hirsutismus, Zyklusstörungen oder Amenorrhoe ein.

CT-Bilder eines adrenalen M. cushing mit Nebennierenadenom links
6 Monate postoperativ



Therapie
Bei der primären Form mit Nebennierenadenom wird dieses laparoskopisch entfernt. Dabei steht sowohl der transabdominelle als auch der retroperitoneale Zugang zur Verfügung. Bei der zentralen Form wird bei Vorliegen eines Hypophysenadenoms dieses operativ entfernt. Da die Hypopyhse in der Sella turcia liegt, kann eine solche Operation transnasal durchgeführt werden.
Hyperaldosteronismus
Das CONN-Syndrom wird durch eine Hyperplasie der Nebennierenrinde ausgelöst, genauer durch eine Hyperplasie der Zona glomerulosa. In ca. 30 % der Fälle ist es somit durch ein Nebennierenadenom ausgelöst. Auch hier gibt es aber extrarenale Formen, wobei ein reninproduzierender Tumor oder eine Nierenarterienstenose ein sekundäres Conn-Syndrom auslösen kann.
Diagnostisch wegweisend sind eine arterielle Hypertonie sowie das gleichzeitige Vorliegen einer Hypokaliämie und Hypernatriämie. In der erweiterten Labordiagnostik lässt sich Aldosteron im Blut und auch im 24-h-Sammelurin nachweisen. Das Renin ist bei der primären Form des Hyperaldosteronismus erniedrigt, bei der sekundären Form erhöht. Zur Klärung der Seitenlokalisation eignet sich am ehesten die MRT und die seitengetrennte Blutentnahme.
Die Therapie besteht aus einer konservativen Behandlung mit einem Aldosteron-Antagonisten wie Spironolacton, eine Indikation zur Operation ergibt sich bei Vorliegen eines Adenoms oder bei Verdacht auf eine Nebennierenkarzinom.
Inzidentalom
Dieser Begriff leitet sich aus der Tatsache ab, dass diese hormoninaktiven Raumforderungen der Nebenniere meist zufällig im Rahmen einer Sonografie der Nieren entdeckt werden. Unter dem Begriff werden sowohl benigne als auch maligne Tumoren zusammengefasst, die im Erwachsenenalter vor allem aus der Nebennierenrinde entstehen können.
Neben dem Ausschluss einer Hormonaktitivät besteht die Diagnostik hautpsächlich in der Lokalisationsfindung, die mittels CT oder kontrastmittelverstärkter MRT erreicht werden kann. Die Indikation zu einer Operation ergibt sich primär aus der Größe eines Befundes. Bis zu 3cm große Inzidenatalome werden alle 6 Monate kontrolliert, ab 5 cm besteht der Verdacht auf ein Nebennierenkarzinom. Sollte sich bei der Operation der Verdacht auf ein Nebennierenkarzinom bestätigen, werden zusätzlich die paraaortalen und parakavalen Lymphknoten ausgeräumt und eine adjuvante Chemotherapie angeschlossen und das Tumorbett bestrahlt.

